Date: Thu, 16 Oct 2014 10:38:59 -0600
Hi,
I know that there was some discussion of this right after the Amber 14
release, but I wasn't sure where we ended up on this.
On Fri, Oct 10, 2014 at 10:25 AM, Jason Swails <jason.swails.gmail.com> wrote:
> Unfortunately, the pmemd.cuda tests have this problem sometimes. The
> larger errors should occur for the stochastic tests (ntt=2 and ntt=3 -- the
> name often tells you if one of those thermostats is involved).
>
> The problem is that the random number generators are different on each GPU
> model. As a result, it was impossible with the Amber 14 code to design a
> test that would give identical results on all GPUs, even if you specified
> the initial seed. As a result, the only cards that all tests pass for are
> the cards that Ross used to create the test files in the first place.
Are we sure this is the correct explanation? I ask because I see some
diffs that don't appear to be related to thermostat. For example, in
the serial CUDA minimization test for chamber/dhfr_pbc at the final
step the dihedral energy absolute diff is 2.33E-01:
cuda/chamber/dhfr_pbc/mdout.dhfr_charmm_pbc_noshake_min.dif
.< DIHED = 739.3609
.> DIHED = 739.3595
This is larger than what is usually considered "acceptable" for CPU -
is it OK for GPU? There are many test diffs (at least using the
compile from the GIT tree as of Oct. 8):
Serial:
89 file comparisons passed
36 file comparisons failed
0 tests experienced errors
MPI:
54 file comparisons passed
33 file comparisons failed
0 tests experienced errors
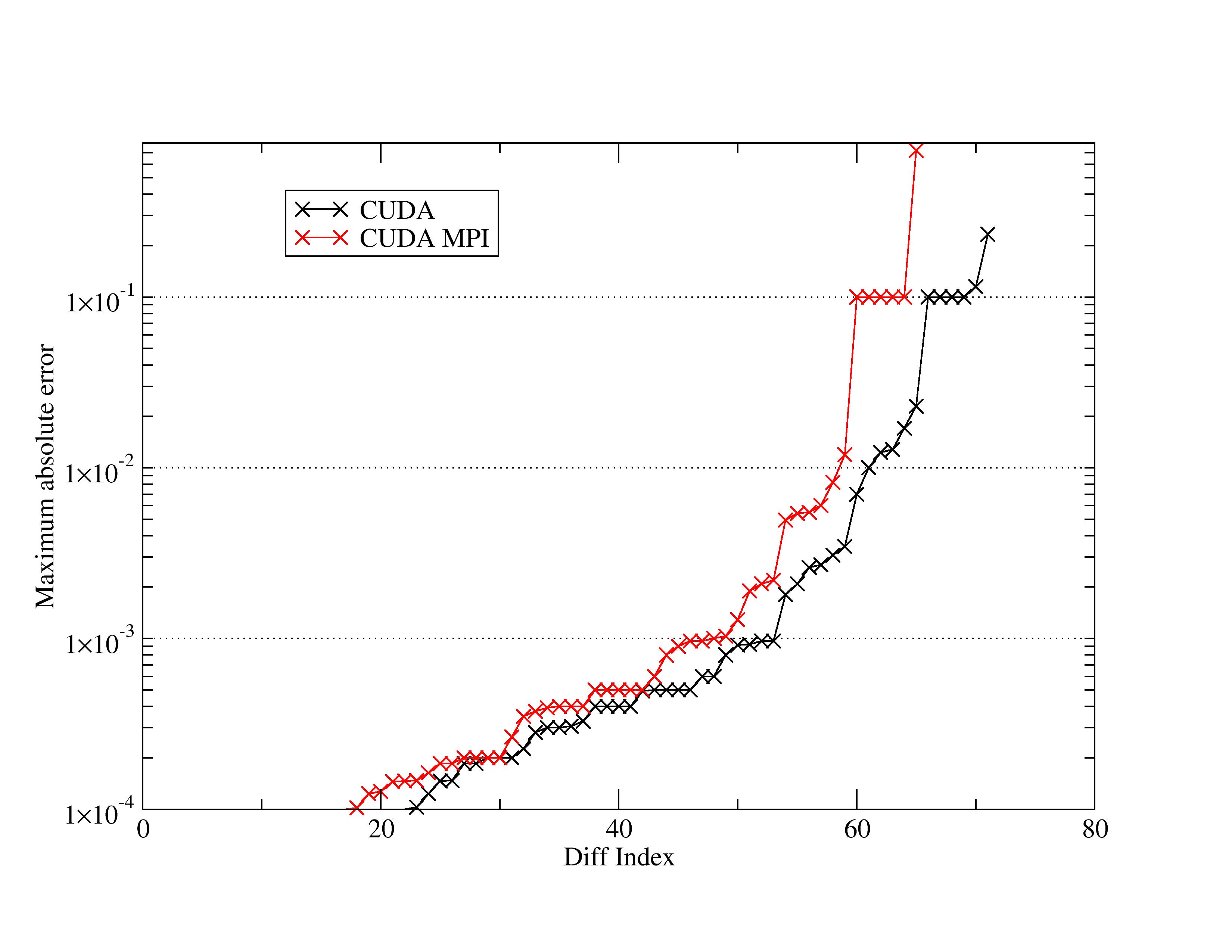
I've attached a plot of the maximum absolute error grabbed from the
related test diff files. From my cursory inspection of the diffs
themselves most of this stuff does appear innocuous. In some cases the
diffs are in the 'RMS fluctuations section', or its only a single step
where e.g. the PRESS variable is off by 0.1, etc. However, I can see
how these results would be very alarming for an everyday user.
If I switch to the DPFP model all serial tests pass, and all parallel
tests pass except for 'cnstph/explicit' (many small differences) and
'lipid_npt_tests/mdout_nvt_lipid14' (1 very small diff in EPtot), so
this does appear to be a precision thing. I'm just wondering if there
isn't some way we can improve the SPFP tests so they "work". I'm
worried that if we get too used to seeing all of these diffs in the
test output it will be harder to spot an actual problem if/when it
arises.
Thoughts?
-Dan
>
> So as long as the diffs appear in some kind of "ntt2" or "ntt3" test
> (Andersen or Langevin thermostats), and the remaining diffs are small, you
> should be fine. FWIW, 37 sounds about the right number to me.
>
> HTH,
> Jason
>
> --
> Jason M. Swails
> BioMaPS,
> Rutgers University
> Postdoctoral Researcher
> _______________________________________________
> AMBER mailing list
> AMBER.ambermd.org
> http://lists.ambermd.org/mailman/listinfo/amber
-- ------------------------- Daniel R. Roe, PhD Department of Medicinal Chemistry University of Utah 30 South 2000 East, Room 307 Salt Lake City, UT 84112-5820 http://home.chpc.utah.edu/~cheatham/ (801) 587-9652 (801) 585-6208 (Fax)
_______________________________________________
AMBER-Developers mailing list
AMBER-Developers.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber-developers
(image/jpeg attachment: Cuda.MaxAbsError.jpg)
