Date: Tue, 4 Apr 2017 11:10:06 -0700
Hello,
I’m running into some problems with energy conservation in an NVE system with pmemd.cuda. I’m running multiple versions of the same system (OPC water with a small molecule) using the same input parameters but different temperatures. Some of these runs show poor energy conservation, heating ~15 K over 50 ns but not all. The same input files run with pmemd.MPI show no obvious heating. I’ve attached plots of the respective output and a tarball with a sample run.
GPU runs were with pmemd.cuda_SPFP. I’ve tried both the fully patch Amber 16 branch, and the master branch.
I’d like to know if there is something I’m doing wrong or if there is a problem with pmemd.cuda_SPFP.
Thank you,
Tyler
_______________________________________________
AMBER-Developers mailing list
AMBER-Developers.ambermd.org
http://lists.ambermd.org/mailman/listinfo/amber-developers
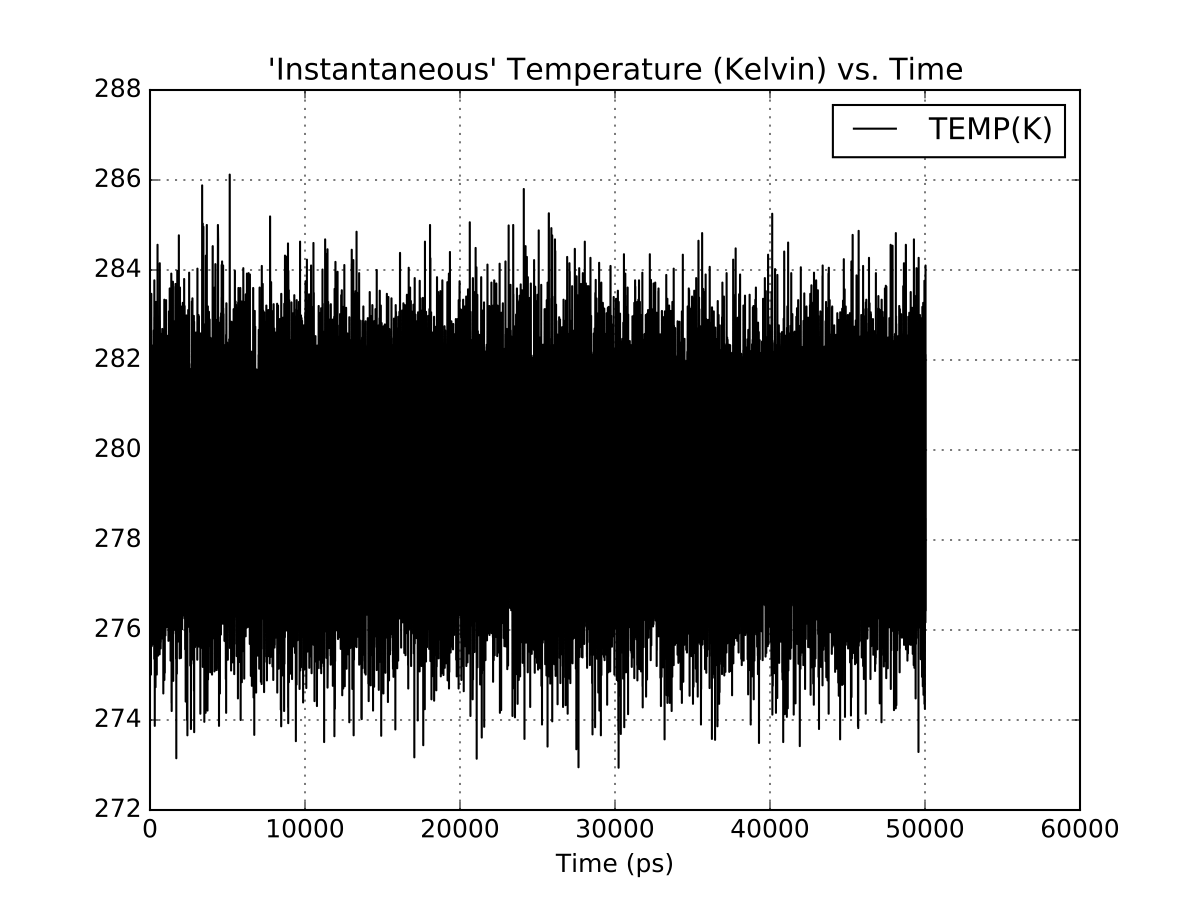
(image/png attachment: cpu-temperature.png)
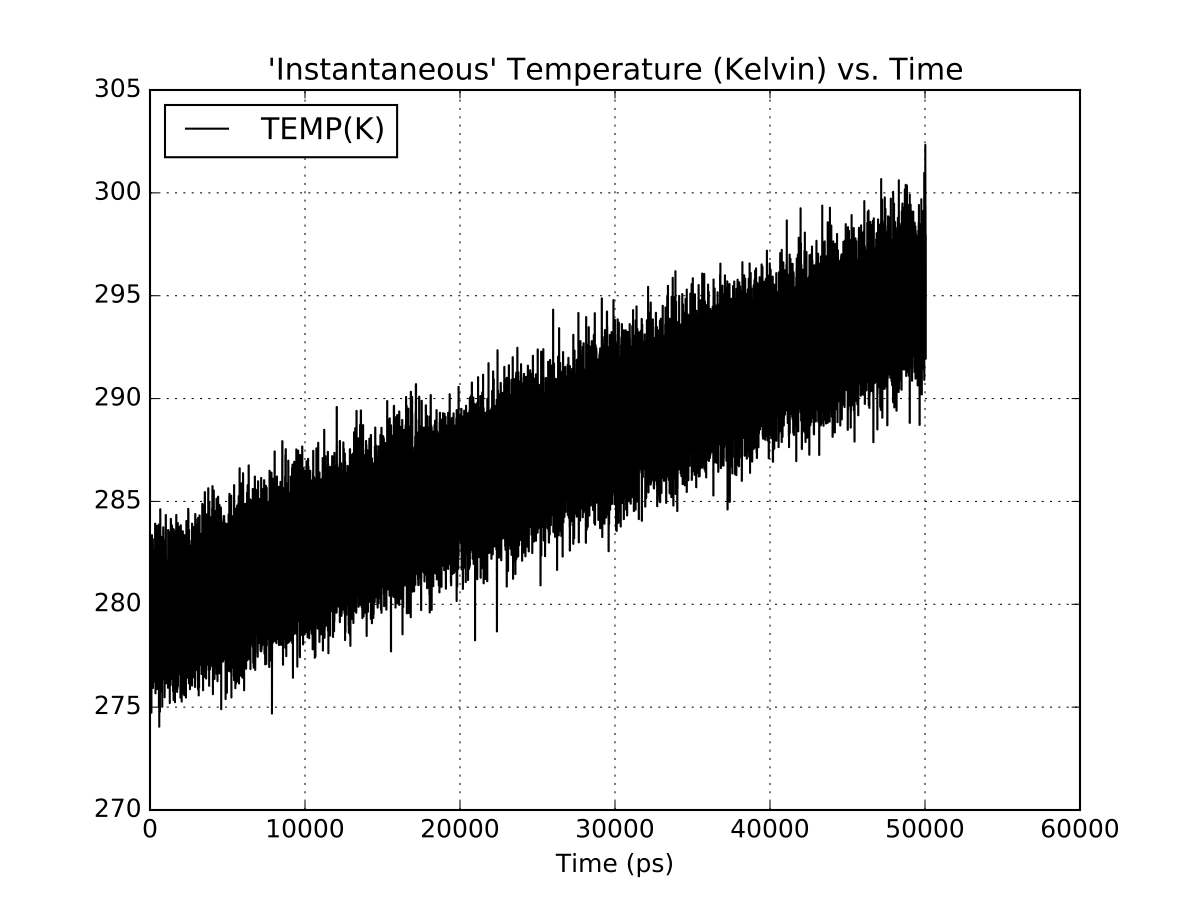
(image/png attachment: gpu-temperature.png)
- application/octet-stream attachment: pDTO_279_tests.tgz
