Date: Wed, 30 Nov 2005 23:48:58 -0700
Dear DAC,
I have been testing the TI approach for a lot of configurations (changing
the parameters like ntc, ntf, etc.) for cytosine to isocytosine and ggcc
to igigicic systems. I have some ideas why this weird results are coming
from. I have attached 3 files taken during the simulation of cytosine to
isocytosine. Let me first explain these images:
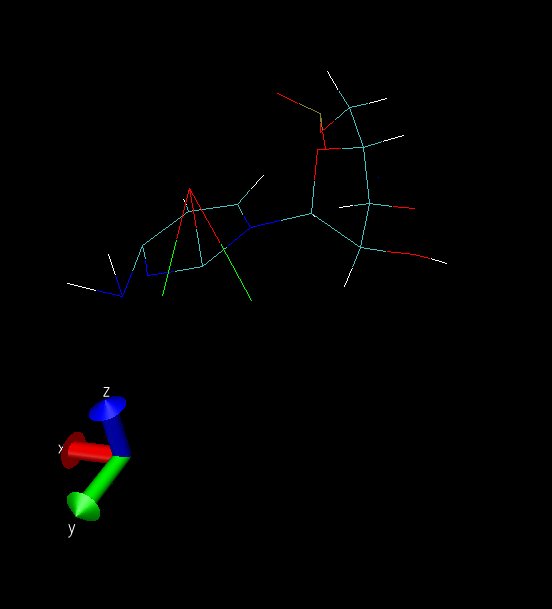
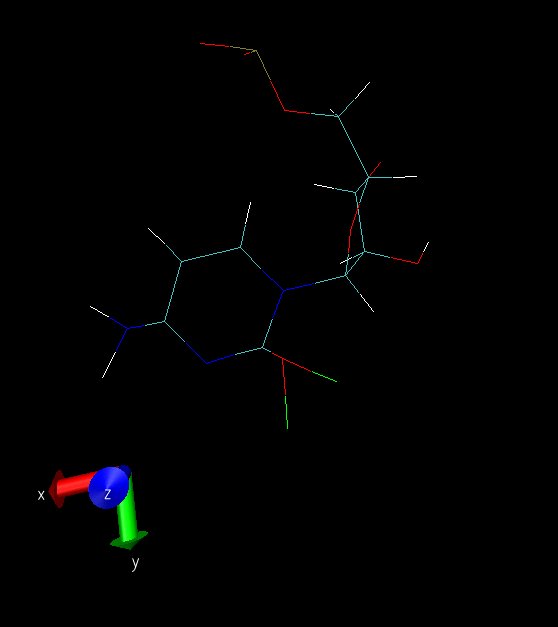
snap01 and snap02 are taken from the same frame from diff. angles. This
simulation is when ntc=1 and ntf=1; SHAKE is OFF. This structure is always
seen in the simulation, namely the amino and carbonyl groups were always
bent like this. The simulation did not blow up, though, and I had a clear
run except for this weird non-planarity of the amino and carbonyl groups
(Note: Those 2 atoms attached to O2 are dummy atoms - red colored).
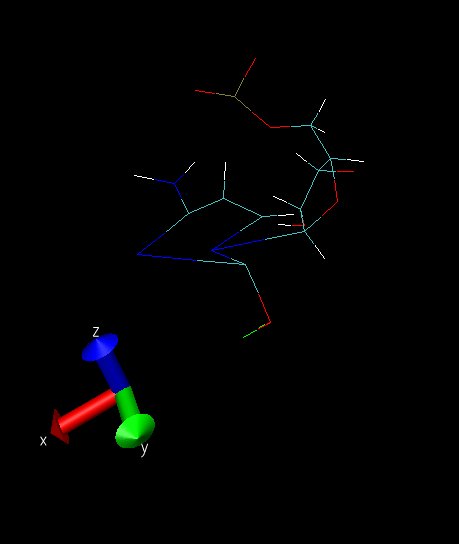
snap03 is a frame from another simulation when ntc=2 and ntf=2, taken
almost at the end of the simulation, before it crashed. As it can be
seen, the ring part is starting to blow up. The simulation crashed, and
in the output file, I had most of the time "vlimit exceeded" messages.
Now, I have two ideas why I am ending up with these crashes.
1. I am thinking that sander is reading the improper torsional parameters
given in frcmod file wrong. Here is the file I am using:
--------------------------- extra.frcmod ------------------------------
GGCC -> iGiGiCiC extra parameters
MASS
DH 1.008
BOND
O -DH 434.0 1.010 [N2-H]
CA-N* 424.0 1.383 [C-N*]
ANGL
DH-O -DH 35.0 120.00 [H -N2-H]
DH-O -C 50.0 120.00 [CA-N2-H]
NA-CA-CB 70.0 111.30 [CB-C -NA]
NA-C -NC 70.0 123.30 [NA-CA-NC]
C -NC-CB 70.0 112.20 [CA-NC-CB]
NC-C -CM 70.0 121.50 [CM-CA-NC]
NC-CA-N* 70.0 118.60 [N*-C -NC]
N2-CA-N* 80.0 120.90 [N*-C -O]
CA-N*-CM 70.0 121.60 [C -N*-CM]
CA-N*-CT 70.0 117.60 [C -N*-CT]
DIHE
DH-O -C -NC 1 0.0 0.0 2. [Dummy Params]
DH-O -C -N* 1 0.0 0.0 2. [Dummy Params]
DH-O -C -CM 1 0.0 0.0 2. [Dummy Params]
C -NC-CA-N* 1 4.80 180.0 2. [X -CA-NC-X]
NC-C -CM-HA 1 2.175 180.0 2. [X -C -CM-X]
NC-C -CM-CM 1 2.175 180.0 2. [X -C -CM-X]
NC-CA-N*-CM 1 1.45 180.0 2. [X -C -N*-X]
NC-CA-N*-CT 1 1.45 180.0 2. [X -C -N*-X]
CA-NC-C -CM 1 4.00 180.0 2. [X -C -NC-X]
CA-N*-CM-H4 1 1.85 180.0 2. [X -CM-N*-X]
CA-N*-CM-CM 1 1.85 180.0 2. [X -CM-N*-X]
CA-N*-CT-OS 1 0.00 0.0 2. [X -CT-N*-X]
CA-N*-CT-H2 1 0.00 0.0 2. [X -CT-N*-X]
CA-N*-CT-CT 1 0.00 0.0 2. [X -CT-N*-X]
N2-CA-N*-CM 1 1.45 180.0 2. [X -C -N*-X]
N2-CA-N*-CT 1 1.45 180.0 2. [X -C -N*-X]
H -N2-CA-N* 1 2.40 180.0 2. [X -CA-N2-X]
CA-NA-C -NC 1 1.35 180.0 2. [X -C -NA-X]
NA-CA-CB-NB 1 3.50 180.0 2. [X -CA-CB-X]
NA-CA-CB-CB 1 3.50 180.0 2. [X -CA-CB-X]
NA-C -NC-CB 1 4.00 180.0 2. [X -C -NC-X]
H -NA-CA-CB 1 1.50 180.0 2. [X -CA-NA-X]
H -NA-C -NC 1 1.35 180.0 2. [X -C -NA-X]
C -NA-CA-CB 1 1.50 180.0 2. [X -CA-NA-X]
C -NC-CB-CB 1 4.15 180.0 2. [X -CB-NC-X]
C -NC-CB-N* 1 4.15 180.0 2. [X -CB-NC-X]
O -C -NC-CB 1 4.00 180.0 2. [X -C -NC-X]
N2-CA-CM-CM 1 2.55 180.0 -2. [X -CA-CM-X]
N2-CA-CM-CM 1 0.0 0.0 3. [Dummy Params]
IMPR
DH-DH-O -C 1.1 180. 2. [X -X -N2-H]
CB-NA-CA-N2 1.1 180. 2. [Dummy Params]
NA-NC-C -O 10.5 180. 2. [X -X -C -O]
CM-C -CM-HA 1.1 180. 2. [X -X -CM-HA]
CM-NC-C -O 10.5 180. 2. [X -X -C -O]
NC-N*-CA-N2 1.1 180. 2. [Dummy Params]
CA-CM-N*-CT 1.0 180. 2. [CM-C -N*-CT]
NONB
DH 1.0000 0.000
--------------------------------------------------------------------------
-- In the IMPR part of the frcmod file, I am stating that I want to keep the carbonyl group planar ( -C-O-(DH)_2 ). I have used exactly the same frcmod file and did a lot of runs in Amber 8 (with different choice of ntc and ntf and etc.), too. I never had any errors or vlimit messages during the simulations, and the simulations ended up with no problem. When I checked out the .traj files of the simulations I ran, the amino group and carbonyl group (with the 2 dummy atoms attached to O2) were jiggling a little bit but I could see that they were trying to stay planar. I wonder if this frcmod file I am using is compatible with AMBER 9. If so, maybe sander is reading the improper terms wrong. 2. My second thought is that the SHAKE algorithm has a bug in it. But still this does not explain why I am ending up with snap01 and snap02 structures (SHAKE is off). Or maybe there is a bug in multisander. But if this was the case, the test simulation I put into /test directory, $AMBERHOME/test/ti_eth2meth, would not work, either. So, I am wondering if it is possible to check whether sander is using the right improper terms or not. I can send a very detailed email for the case cytosine->isocytosine transformation, but I would like to know, first, if the above frcmod file is compatible with Amber 9. Or if it is possible to check whether sander is using the right improper terms. Best, PS 1: All structures were neutralized first with Na+ ions and solvated in water. I have created my own .prepi files for the nucleic acids and used these files in xleap to create the parm/top files. PS 2: I did not run any equilibration neither in amber 9 not amber 8 simulations. I have used the following input file (and I was changing ntc, ntf, irest, ntx, icfe, etc. in the simulations). I have kept the "tempi = 300.0, temp0 = 300.0" part in all simulations. I did some heating simulations before, and they were also giving the same vlimit messages in the output file. So, I decided to use "tempi = 300.0, temp0 = 300.0" all the time. --------------------------- mdin ----------------------------------- TEST MD &cntrl imin = 0, irest = 1, ntx = 5, ntb = 1, cut = 8, ntr = 0, ntc = 2, ntf = 2, tempi = 300.0, temp0 = 300.0, ig=233, ntp=0,taup=2.0,pres0=1, ntt = 3, gamma_ln = 1.0, nstlim = 10000, dt = 0.001, nscm = 1000, nrespa=1, ntpr = 100, ntwx = 10, ntwr = 1000, icfe=$icfe, klambda=6, clambda=$lambda / -------------------------------------------------------------------- On Tue, 29 Nov 2005, David A. Case wrote: > On Tue, Nov 29, 2005, Ilyas Yildirim wrote: > > > icfe=2, klambda=6, clambda=0.5 > > ... > > > vlimit exceeded for step 218 ; vmax = 22.6949147823007 > > > > Coordinate resetting (SHAKE) cannot be accomplished, > > > > I am not sure why I am ending up with this kind of an error. I > > first thought that the modified .frcmod might be the reason, but this > > mod.frcmod file is working fine with AMBER 8. I have checked out the > > .mdcrd trajectories, and the -NH2 and -O-(DH)2 parts of the structure is > > doing really weird stuff. > > Have you run long-ish equilibrations at both endpoints? You should always do > this first. Look at the trajectories visually...see if you can find out what > the "really weird stuff" is coming from. > > How big are the values of DV/DL that you get? Big values indicate that a > single geometry cannot simultaneously accommodate both endpoint potentials. > Ideally, you want the optimized geometries of the two endpoints to be pretty > similar to one another. Another obvious test is to set icfe=klambda=1, to > make sure that there is not something arising from the new code you added. > > ...dac > > -- Ilyas Yildirim --------------------------------------------------------------- - Department of Chemisty - - - University of Rochester - - - Hutchison Hall, # B10 - - - Rochester, NY 14627-0216 - Ph.:(585) 275 67 66 (Office) - - http://www.pas.rochester.edu/~yildirim/ - ---------------------------------------------------------------
(IMAGE/jpeg attachment: snap01.jpg)
(IMAGE/jpeg attachment: snap02.jpg)
(IMAGE/jpeg attachment: snap03.jpg)
